Unit 2: Compound Structure and Properties
This unit connects what atoms are (Unit 1) to how they assemble into compounds and what that assembly predicts about structure, bonding, and bulk properties. We will move from qualitative pictures of electrons in bonds to rules for naming and drawing molecules, then to three-dimensional molecular geometry and the orbital picture that supports it.
Types of chemical bonding
Electrons are the currency of chemical bonding. Whether electrons are fully transferred, shared, or pooled determines the type of bond and, in broad terms, how strong and how directional that interaction is.
Ionic bonding
An ionic bond arises between a metal and a nonmetal when electrons are transferred so that both partners approach stable electron counts. The metal loses electrons to become a cation and the nonmetal gains electrons to become an anion. The resulting ions attract one another by Coulomb’s law (see Unit 1). For two point charges \(Q_1\) and \(Q_2\) separated by distance \(r\), the electrostatic potential energy has the familiar form as seen in Unit 1 (with \(k\) a constant):
\[U = k \frac{Q_1 Q_2}{r}\]Oppositely charged ions (\(Q_1 Q_2 < 0\)) lower their energy as \(r\) decreases, which is why a crystal lattice of alternating cations and anions is stable. Lattice energy trends (larger charges, smaller ions) follow from this same inverse dependence on \(r\) and linear dependence on the product of charges.
A standard example is sodium chloride: chlorine accepts an electron to form \(\text{Cl}^-\), and sodium becomes \(\text{Na}^+\). The compound is held together by attraction between those ions, not by shared pairs in the sense of a covalent bond. However, counterintuitively, the ionic bond is the strongest type of bond.
Covalent bonding
A covalent bond forms between atoms that are both relatively electronegative, typically two nonmetals, when they share one or more pairs of valence electrons. Neither atom fully owns the electrons; instead, both nuclei are attracted to the same shared density. Bond polarity appears when the two atoms differ in electronegativity: the shared pair is pulled toward the more electronegative atom, giving partial charges (\(\delta^+\) and \(\delta^-\)) even though the bond is still covalent. Usually, the cutoff for a bond being polar is a absolute difference of electronegativity above 0.5, and for an ionic bond it is usually 2.0.
Compared with ionic lattices, individual covalent bonds are often discussed in terms of bond energy (the energy required to break one mole of that bond in the gas phase, for a defined process). Triple bonds are generally stronger and shorter than double bonds, which are stronger and shorter than single bonds between the same elements.
Metallic bonding
In a metal, valence electrons are not localized between two nuclei. Instead, they occupy a sea of delocalized electrons that extends through the crystal, while the metal cations sit in orderly positions. That delocalization explains electrical conductivity and malleability: applying stress shifts ion layers without necessarily breaking localized bonds, because the electron sea can still bind the structure.
Nomenclature
Systematic names tell a chemist which elements are present, in what ratio, and (for ions) what the charges are. The rules differ for ionic compounds and molecular (covalent) compounds.
Molecular compounds
If a substance contains only covalent bonds between nonmetals, it is a molecular compound. For a binary molecular compound (exactly two different elements), use numerical prefixes (mono-, di-, tri-, …) to indicate how many atoms of each element are present. The less electronegative element is usually named first; the second element takes an -ide ending. For example, \(\text{N}_2\text{O}\) is dinitrogen monoxide, the prefixes di- and mono- describe a two-to-one ratio of nitrogen to oxygen.
If the second species is a polyatomic ion, name it as such rather than forcing an -ide ending on a single atom. Acids containing hydrogen often have special names; those conventions are developed in Unit 8.
Ionic compounds
An ionic compound is named beginning with the cation. For metals that can take more than one charge in compounds (many transition metals) include the charge as a Roman numeral in parentheses after the metal name: \(\text{Fe}^{3+}\) in a compound is iron(III). Then name the anion: monatomic anions use the -ide ending (\(\text{F}^-\) is fluoride). Ammonium, \(\text{NH}_4^+\), behaves like a polyatomic cation in naming even though it contains no metal. Hydrogen ion, \(\text{H}^+\), does not behave like a typical metal cation in nomenclature. Prefixes are not used to show stoichiometry in simple ionic names because the charges determine the ratio.
Organic molecules
Organic chemistry is the study of compounds built around carbon frameworks. Many of the same ideas (Lewis structures, hybridization, geometry) carry over, but carbon compounds also use a parallel naming system (IUPAC names, functional groups, and common names). Those details are not necessary for AP Chemistry, and usually organic molecules will be given to you, or will show up on the list of polyatomic ions.
Drawing two-dimensional structures
Lewis structures are schematic: they show valence electrons and bonds in two dimensions even though real molecules are three-dimensional. They are still very useful for drawing bonds, assigning formal charge, and spotting resonance.
The octet rule
The octet rule says that in many compounds, atoms are most stable when they are surrounded by eight valence electrons (four pairs), matching the noble gas configuration of the noble gas in the same period. Hydrogen and helium are exceptions in the strict sense: hydrogen/helium aim for two electrons (a duet), not eight.
Some period 2 elements (notably boron, beryllium, and aluminum in compounds such as \(\text{BF}_3\)) may be electron-deficient, with fewer than eight valence electrons on the central atom in a Lewis structure that still matches experiment. Specifically, they aim to have twice the number of valence electrons they currently have in their shell.
Heavier main-group elements (the square from phosphorus through astatine in the \(p\) block, in the usual textbook treatment) can exhibit expanded octets, using \(d\) orbitals in the hybridization picture to accommodate more than eight valence electrons when they are the central atom in certain compounds.
When drawing, always ask whether the central atom is allowed to expand or must remain octet-complete for the story you are telling.
Formal charge
Formal charge is a bookkeeping tool for a single Lewis structure. It assigns each valence electron in the structure either to an atom or to a bond. For an atom,
\[\text{FC} = (\text{valence } e^- \text{ for the free atom}) - (\text{nonbonding } e^-) - \frac{1}{2}(\text{bonding } e^-)\]Structures that minimize formal charge (and place negative formal charge on more electronegative atoms when a choice exists) are generally preferred as major contributors in a resonance hybrid.
Resonance
Resonance occurs when two or more Lewis structures differ only in the placement of \(\pi\) bonds and lone pairs, not in the arrangement of nuclei. The classic example is nitrate, \(\text{NO}_3^-\): the double bond can be drawn to any of the three oxygen atoms with equal validity. The real ion is a resonance hybrid: a single averaged distribution in which N–O bonds are equivalent by symmetry, with fractional bond order between single and double.
For resonance to be meaningful, the alternative structures must involve the same connectivity; swapping which atoms are bonded (for example, interchanging roles of carbon and nitrogen) is not resonance.
Constructing Lewis structures
A practical algorithm:
- Count total valence electrons (adjust for charge on ions).
- Choose a central atom—usually the least electronegative element that is not hydrogen. For only two atoms, there is no unique “center”; place them adjacent.
- Connect atoms with single bonds first, then distribute remaining electrons as lone pairs to satisfy the octet rule (or duet for H) on outer atoms.
- If the central atom lacks an octet, form multiple bonds from outer atoms that can accept higher bond order (often O, N, or C), respecting the octet / expansion rules above.
- If several arrangements remain, favor the one with formal charges closest to zero.
- Indicate resonance with double-headed arrows between structures.
- Put brackets around the compound if it has a charge.
VSEPR theory
Valence-shell electron-pair repulsion (VSEPR) theory explains electron-domain geometry and molecular shape by assuming that both bonding pairs and lone pairs around a central atom repel one another. The arrangement that maximizes separation minimizes repulsion (thus minimizes the amount of energy needed to hold the molecules) and thus corresponds to observed geometry. A double bond or triple bond counts as one electron domain toward the steric number, just like a single bond or a lone pair.
Typical pairings of steric number and lone pairs give names such as linear, trigonal planar, tetrahedral, trigonal bipyramidal, and octahedral for the electron-domain geometry; lone pairs then influence the molecular geometry (for example, bent instead of trigonal planar when one lone pair sits on a central atom with three domains).
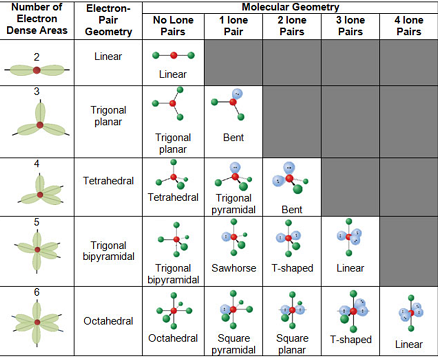
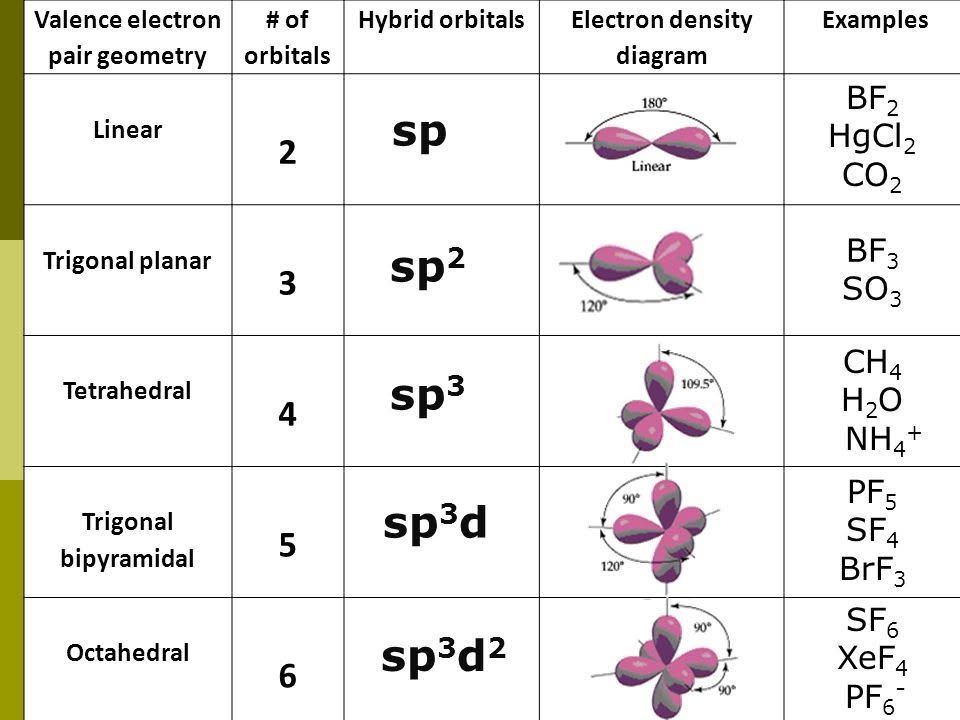
Hybridization
Atomic orbitals on a bonded atom can mix to form hybrid orbitals that are consistent with VESPR. For carbon in many organic molecules, four sigma frameworks point toward the corners of a tetrahedron, described by \(\text{sp}^3\) hybridization. \(\text{sp}^2\) hybrids lie in a plane at \(120^\circ\) (trigonal planar arrangement); \(\text{sp}\) hybrids are linear at \(180^\circ\). In addition, \(\text{sp}^3 d\) hybridization occurs in trigonal bipyramidal configurations, while \(\text{sp}^3 d^2\) hybridization occurs in an octahedral configuration.
Sigma bonds, pi bonds, and bond order
A sigma bond (\(\sigma\)) has electron density concentrated along the internuclear axis; it arises from head-on overlap of hybrid or atomic orbitals. A pi bond (\(\pi\)) forms from side-by-side overlap of unhybridized \(p\) orbitals above and below the sigma framework. A single bond is one \(\sigma\); a double bond is one \(\sigma\) plus one \(\pi\); a triple bond is one \(\sigma\) plus two \(\pi\).
Bond order is half the difference between bonding and antibonding electrons in molecular orbital theory, but in Lewis terms it is simply the average number of bonding electron pairs between two atoms across resonance structures. Bond length decreases and bond strength increases as bond order increases between the same two elements.
Potential energy and bond formation
When two atoms approach, the potential energy of the system typically drops as attractive interactions dominate, passes through a minimum at an equilibrium bond length, and then rises steeply as nuclear repulsion dominates at short distance.
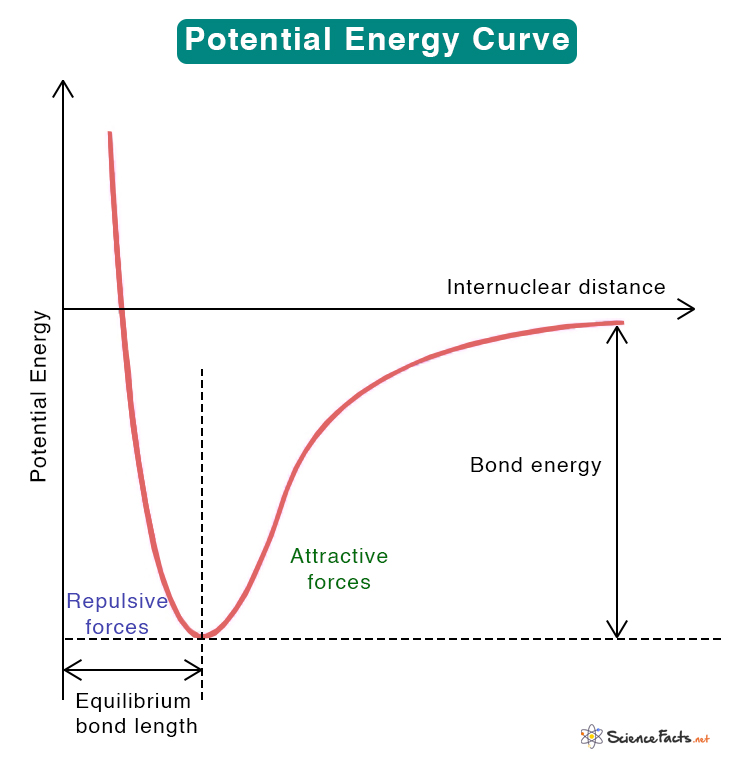
Bond energy is related to the depth of that well. Comparing curves for the same bond order (single vs double vs triple) illustrates why higher bond order correlates with shorter, stronger bonds.
These same curves also motivate reaction coordinates later: along a reaction path, the system moves on a potential energy surface connecting reactants, transition states, and products.
The Born–Haber cycle
First, before we begin, here are some important terms:
- \(U_{lattice}\) = Lattice Energy
- \(\Delta U\) = Change in Potential Energy
- \(\Delta H\) = Change in Enthalpy
- \(\Delta H_{\text{ea}}\) = Enthalpy of Electron Gain
- \(IE\) = Ionization Energy
- \(D_{A-B}\) = Bond Enthalpy between A and B
The Born–Haber cycle is a Hess’s law (See more in Unit 6) construction for an ionic solid. It expresses the standard enthalpy of formation \(\Delta H_f^\circ\) of the compound from its elements in standard states as a sum of steps that convert those elements into gas-phase ions and then let those ions crystallize. Any one unknown step (most often lattice energy) can be found if the others are known.
Take a 1:1 alkali halide \(\text{MX}\), formed from \(\text{M}(s)\) and \(\tfrac{1}{2}\text{X}_2(g)\). Imagine the path:
-
Atomize the metal (sublimation): \(\text{M}(s) \rightarrow \text{M}(g)\) with \(\Delta H = \Delta H_{\text{sub}}\) (usually endothermic).
-
Ionize the metal: \(\text{M}(g) \rightarrow \text{M}^+(g) + e^-\) with \(\Delta H = \text{IE}\) (endothermic; use the correct successive ionization energies if more than one electron is lost).
-
Atomize the halogen: \(\tfrac{1}{2}\text{X}_2(g) \rightarrow \text{X}(g)\) with \(\Delta H = \tfrac{1}{2}D_{\text{X–X}}\) (endothermic; half the \(\text{X–X}\) bond enthalpy).
-
Attach an electron to the halogen: \(\text{X}(g) + e^- \rightarrow \text{X}^-(g)\) with \(\Delta H = \Delta H_{\text{ea}}\). For halogens this step is exothermic, so \(\Delta H_{\text{ea}}\) is negative when reported as an enthalpy change. (If a table lists electron affinity with a different sign convention, convert it to \(\Delta H\) for this step before you add.)
-
Form the crystal from gas ions: \(\text{M}^+(g) + \text{X}^-(g) \rightarrow \text{MX}(s)\). This step is strongly exothermic. Textbooks often define lattice energy \(U_{\text{lattice}}\) as a positive number equal to the endothermic enthalpy of the reverse process: one mole of solid separated into isolated gaseous ions:
Then step 5 (lattice formation from ions) has \(\Delta H = -U_{\text{lattice}}\).
Because the overall enthalpy change from \(\text{M}(s) + \tfrac{1}{2}\text{X}_2(g)\) to \(\text{MX}(s)\) is \(\Delta H_f^\circ\),
\[\Delta H_f^\circ(\text{MX},\,s) = \Delta H_{\text{sub}} + \text{IE} + \frac{1}{2}D_{\text{X–X}} + \Delta H_{\text{ea}} - U_{\text{lattice}}.\]Rearranging isolates the lattice term:
\[U_{\text{lattice}} = \Delta H_{\text{sub}} + \text{IE} + \frac{1}{2}D_{\text{X–X}} + \Delta H_{\text{ea}} - \Delta H_f^\circ.\]For salts with other stoichiometries (e.g. \(\text{MgCl}_2\), \(\text{Na}_2\text{O}\)), use the correct multiple of atomization, all required ionization steps, the appropriate nonmetal atomization (e.g. \(\tfrac{1}{2}\text{O}_2\)), and electron-gain steps that match the anion charge (second-electron addition to oxygen is very endothermic; the huge lattice energy of oxides is what makes the overall formation from elements favorable). The cycle is still closed: the sum of steps along one route equals the sum along any other route between the same two thermodynamic states.
Qualitatively, larger ion charges and smaller ions (shorter internuclear distances in the lattice) increase \(U_{\text{lattice}}\), consistent with Coulomb attraction in the lattice and with the trends introduced earlier in this unit. A picture has been added below for illustrative purposes.